|
Článek
|
Příčina
|
Korekce
|
1.1
|
Systém nebo chromatografická kolona není dostatečně ekvilibrována
(zejména při chromatografii iontových párů).
|
Stabilizace (ekvilibrace) celého systému (kolona a detektor), zejména pokud se jedná o chromatografii iontových párů (IPC), celkový objem solventu k ekvilibraci celého systému je asi 30-100 ml při průtoku 1 ml/min. Problém se může projevit zejména při přechodu ze systému normální fáze na reverzní fázi a nedostatečném propláchnutí celého systému. Při proplachu systému je nutné vždy používat taková po sobě jdoucí rozpouštědla, která jsou vzájemně mísitelná. Při použití pufrů jako mobilní fáze je nutné provést vždy důkladné vymytí pufrů ze systému a ověřovat rozpustnost přidávaných pufrů v mobilní fázi, aby nedošlo při míchání složek mobilní fáze na pumpě i v systému k vysrážení nerozpustných anorganických či organických solí. V případě, že dojde k vysrážení nerozpustných solí v systému je nutné celý systém po vyřazení analytické kolony proplachovat vodou nebo 1 molární kyselinou dusičnou, vodou a nakonec čistým methanolem.
|
1.2
|
Kontaminovaná nebo degradována kolona nebo předkolona.
|
K indikaci tohoto problému je nutné:
- vyměnit kolonu za novou kolonu toho samého typu
- ekvilibrovat kolonu minimálně 10 kolonovými objemy mobilní fází
- monitorovat základní linii
Jestliže problém není odstraněn, kolonu je možné propláchnout podle jednoho z dekontaminačních programů a pokusit se o její oživení nebo je nutné ji vyměnit.
Dekontaminační program chromatografické kolony je uveden zde.
Pro dekontaminaci proteinových kontaminantů je možno použít nástřik 200 µl dimethylsulfoxidu (DMSO) na kolonu do mobilní fáze se kterou je DMSO mísitelný. Tento postup se může opakovat dva až třikrát.
Při použití nové kolony je vhodné zaznamenat tlak na chromatografické koloně za daných podmínek. Indikace kontaminace nebo degradace kolony je pak velmi rychlá, neboť stárnutím kolony dochází ke zvýšení tlakového spádu na koloně.
|
1.3
|
Netěsnost v systému (dávkování vzorku, cela detektoru, spoje kapilár).
|
Zkontrolovat všechny spoje celého systému, popřípadě vyměnit všechny ferullky. Nejlepší
postup k detekci netěsnosti v systému je za použití suchého filtračního
papíru, který za průtoku mobilní fáze se přiloží ke každému spoji a zkontroluje
se, zda se neobjeví mokrá skvrna.
|
1.4
|
Mobilní fáze je kontaminována,popřípadě je špatně odvzdušněna.
|
Je-li podezření na kontaminaci mobilní fáze, použije se čerstvě připravená mobilní fáze. K prevenci je vhodné používat vždy rozpouštědla co nejvyšší čistoty (HPLC grade) a všechny připravené mobilní fáze by se měly filtrovat přes filtr 0,45 µm. K odvzdušnění mobilní fáze je možno použít evakuaci mobilní fáze před vlastním použitím, nebo použít heliový nebo vakuový degaser.
|
1.5
|
Kontaminovaný in-line filtr mobilní fáze nebo zásobník mobilní fáze.
|
Použít nový, čistý in-line filtr mobilní fáze a zásobník mobilní fáze. Vyčistit in-line filtr mobilní fáze – viz rovněž článek tabulky 2.1
|
1.6
|
Mobilní fáze obsahuje stabilizátor.
|
Jako nejčastější stabilizátor se používá azid sodný, který zabraňuje mikrobiologickému růstu v mobilní fázi (zejména mobilní fáze obsahující fosfátový pufr). Je-li podezření na tuto příčinu, použije se čerstvě připravená mobilní fáze bez stabilizátorů.
|
1.7
|
Kontaminovaná cela detektoru.
|
Je-li podezření na kontaminaci cely detektoru, musí se cela detektoru pročistit:
- v případě použití mobilní fáze s pufrem, propláchnout celu detektoru vodou, methanolem a následně opět vodou
- v případě použití nepolární mobilní fáze, propláchnout celu detektoru rozpouštědlem tetrahydrofuran+voda (50 + 50)
- jestliže tento problém přetrvává, použije se proplach 6 M-HNO3, vodou a následně methanolem
Při této proceduře je nutné brát ohled na tlakový limit cely detektoru. Proplachovací kapalina se vždy nasává, inkdy se netlačí do cely detektoru!
|
1.8
|
Nekorektní nastavení vlnové délky detektoru.
|
Změnit nastavení vlnové délky, která je vhodnější pro danou použitou mobilní fázi (cutoff mobilní fáze).
|
1.9
|
Teplotní změny v systému na chromatografické koloně).
|
Je-li podezření na teplotní fluktuaci použije se temperace chromatografické kolony tak, aby se teplota kolony pohybovala asi 5 °C nad teplotou okolí v rozmezí teplot ± 2 °C
|
|
Článek
|
Příčina
|
Korekce
|
2.1
|
HPLC pumpa pulsuje (krátké vlny).
|
Jako možné příčiny mohou být následující:
- pumpa není primována nebo je přítomna vzduchová bublinka v hlavě pumpy a v obou případech je nutné primovat pumpu
- in-line filtr mobilní fáze je částečně blokován a je nutný je pročistit – vyjmout filtr, 5 minut čistit v 6 M-HNO3 v ultrazvuku, následně třikrát propláchnout vodou a methanolem a profouknout dusíkem k suchu
- podtéká hlava pumpy a je nutná výměna těsnění pístu
|
2.2
|
Mobilní fáze není dokonale promíchaná (gradient).
|
Viz článek tabulky 4.4
|
2.3
|
Teplotní fluktuace na chromatografické koloně (dlouhé vlny).
|
Viz článek tabulky 1.9
|
2.4
|
Mobilní fáze je recyklována a vrácena zpět z detektoru na chromatografickou kolonu (dlouhé vlny).
|
Jestliže to není absolutně nutné, vyhnout se tomuto řešení a nepoužívat recyklovanou mobilní fázi a vždy používat filtrovanou a čerstvou mobilní fázi.
|
|
Článek
|
Příčina
|
Korekce
|
 3.1 3.1
|
Vzduchová bublinka v cele detektoru.
|
Vzduchovou bublinku je nutno odstranit a to zvýšením průtoku mobilní fáze nebo nepatrným zvýšením zpětného tlaku na výstupu z detektoru (ucpání kapiláry na několik sekund).
Je nutné mít na paměti tlakový limit cely detektoru.
Pro prevenci tvorby vzduchových bublinek v cele detektoru je vhodné použití vakuového degaseru a zapojení kapiláry 30 až 90 cm o vnitřním průměru 0,23 mm na výstup detektoru. Tato kapilára plní funkci tlakového omezovače a 90 cm této kapiláry zvýší tlak o 200 až 300 kPa při průtoku mobilní fáze 1 ml/min.
|
3.2
|
Netěsnost v cele detektoru.
|
Ke kontrole tohoto problému je nutné odstranit pouzdro detektoru a není-li netěsnost viditelná, je nutné učinit následující:
- propláchnout celu detektoru methanolem
- připojit zdroj helia nebo dusíku na vstup cely detektoru a propláchnout plynem k suchu
- jestliže se příčina ztratí, existuje netěsnost uvnitř cely detektoru
|
3.3
|
Nastavení vysoké citlivosti detektoru.
|
Nastavit nižší citlivost detektoru.
|
|
Článek
|
Příčina
|
Korekce
|
4.1
|
HPLC pumpa pulsuje (krátké vlny).
|
Viz článek tabulky 2.1
|
4.2
|
Systém nebo chromatografická kolona není dostatečně ekvilibrována (zejména pro chromatografii iontových párů).
|
Viz článek tabulky 1.1
|
4.3
|
Teplotní fluktuace na chromatografické
koloně (dlouhé vlny).
|
Viz článek tabulky 1.9
|
4.4
|
Mobilní fáze není dokonale promíchaná (gradient).
|
Ke potvrzení této příčiny je nutný následující postup:
- mobilní fází, která je ručně namíchaná, filtrovaná a odvzdušněná se ekvilibruje chromatografická kolona minimálně 5 až 10 kolonovými objemy
- na kolonu se minimálně 3 krát nanese standard a retenční čas se srovná s předchozími retenčními časy
- je-li retenční čas reprodukovatelný pak je indikován problém míchání mobilní fáze
K prevenci tohoto problému je vhodné umístit na výstup pumpy a vstup dávkovače vzorku kapiláru délky 150 až 300 mm o vnitřním průměru 1 mm. Zařazením této kapiláry do systému se zvýší minimálně mrtvý objem systému a ani nedochází k distorzi píků. Zvýší se laminární proudění uvnitř kapiláry a doba zdržení.
Jestliže nedojde k odstranění problému, je nutné umístit dynamický nebo statický mixer mezi detektor a dávkovač vzorku.
|
4.5
|
Kontaminovaná nebo degradována kolona nebo předkolona.
|
Viz článek tabulky 1.2. V případě, že přetrvává tento problém, je možnou příčinou kontaminovaná
mobilní fáze.
|
|
Článek
|
Příčina
|
Korekce
|
6.1
|
Nesprávné nastavení průtoku mobilní fáze.
|
Kontrola nastavení průtoku mobilní fáze na HPLC pumpě.
|
6.2
|
Špatná mobilní fáze.
|
Kontrola správnosti složení mobilní fáze.
|
6.3
|
Jiný typ kolony nebo velikost
(délka, průměr).
|
Kontrola typu a rozměrů kolony (délka, průměr), kontrola, zda je kolona zapojena ve směru toku mobilní fáze.
|
6.4
|
Špatné nastavení teploty při temperaci
chromatografické kolony.
Změna teplotních podmínek na koloně.
|
Kontrola nastavení teploty kolony na teplotním modulu. Použít temperaci chromatografické kolony.
|
6.5
|
Kontaminovaná nebo degradována kolona nebo předkolona.
|
Viz článek tabulky 1.2
|
6.6
|
Změněna průtoku mobilní fáze
|
Kontrola nastavení průtoku mobilní fáze – nastavit příslušný průtok mobilní fáze (nejlépe 1 ml/min) a k indikaci problému udělat následující:
- na výstup detektoru umístit odměrný válec objemu 5 až 10 ml
- po dobu 4 až 8 minut měřit objem mobilní fáze proteklé systémem
- vypočítat aktuální průtok mobilní fáze
Přetrvává-li problém, došlo k poškození čerpadla, nutný zásah servisního technika.
|
|
Článek
|
Příčina
|
Korekce
|
7.1
|
Kontaminovaná nebo degradovaná kolona nebo předkolona nebo kontaminován in-line filtr mobilní fáze.
|
Příčinou vzniku dvojitého píku mohou být mikrokanálky, které vznikají stárnutím kolony a to zejména na počátku kolony. V těchto kanálcích pak dochází ke stagnaci mobilní fáze a separující se látky a dochází pak ke vzniku dvojitého píku. Problém lze odstranit otočením chromatografické kolony proti toku mobilní fáze a kolonu používat v tomto módu – kanálky se pak nachází na konci kolony, kdy jsou látky již separovány. Takto používaná chromatografická kolona nemá však příliš dlouhou životnost. Další korekce viz tabulka č.1.
|
7.2
|
Příliš vysoká nástřiková hmotnostní koncentrace (objem nástřiku /µl/ nebo koncentrace /µg/ml). Nekompatibilní solvent vzorku s mobilní fází.
|
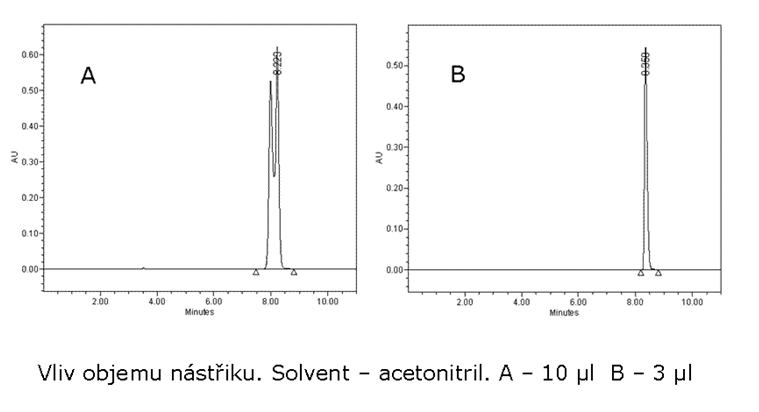
Při vysoké nástřikové hmotnosti se dostáváme do nelineární oblasti absorpční isotermy, chromatografická kolona přestává separovat. Je nutné naředit vzorek nebo snížit objem dávkovaného vzorku na chromatografickou kolonu. Příčina může také být v solventu vzorku. Použít „slabší“ solvent (solvent o menší eluční síle než původní solvent např. místo acetonitrilu použít methanol). Vliv objemu nástřiku na separaci je ukázán na obrázku č. 7.1. Viz článek tabulky č. 8.4
|
7.3
|
Špatný výběr separačního systému.
|
Tento problém se objevuje zejména při separaci látek iontové povahy. Při výběru separačního systému (mobilní fáze) kdy pH = pK separované látky a v systému jsou v rovnováze nedisociovaný a disociovaný podíl látky. Situaci v systému vystihuje obrázek č. 7.2.
|
7.4
|
Sekundární ekvilibrace
|
U některých solutů dochází k přechodu z jedné formy na druhou, zejména v závislosti na pH prostředí. Tento fenomén je nejlépe popsán při analýze cukrů, dalším typickým příkladem jsou aldehydy a ketony (isomerie, která se nazývá enolizace). V případě, že rychlost reakce je vysoká, většinou problémy nenastanou, v případě pomalé reakce dochází k distorzi píku. Za normálních podmínek je u enolizace rovnováha posunuta na levou stranu ve prospěch keto-formy (disociace vazby uhlíku s vodíkem je katalyzována zejména zásadami). Chromatogram separace a vliv pH mobilní na tvorbu keto-enol formy solutu. pH mobilní fáze 7.3.
|
7.5
|
Netěsnost v systému (vstup na kolonu)
|
V případě netěsnosti v systému (vstup na kolonu) dochází k podtékání mobilní fáze a může to být doprovázeno poklesem tlaku v systému a posunem retenčního času. Chromatogram (pík) pak může mít tento tvar (obrázek 7.4).
|
|
Článek
|
Příčina
|
Korekce
|
8.1
|
Kontaminovaná nebo degradována
kolona nebo předkolona nebo kontaminován in-line filtr mobilní fáze.
|
Viz článek tabulky 7.1
|
8.2
|
Příliš vysoká nástřiková hmotnostní koncentrace (objem nástřiku /µl/ nebo koncentrace /µg/ml).
|
Viz článek tabulky 7.2
|
8.3
|
Práce v nelineární oblasti absorpční isotermy.
|
Nástřiková koncentrace je nízká, přesto se dostaneme do nelineární oblasti absorpční isotermy. V případě nelineární
konkávní isotermy se zvyšující se nástřikovou koncentrací se zvyšuje retenční čas tak, jak je ukázáno na obrázku č.9, v případě nelineární konvexní isotermy je tomu přesně naopak – se zvyšující se nástřikovou koncentrací se retenční čas snižuje. Platí zejména pro GC. Názorný vliv adsorbce na retenci je ukázán na obrázku.
|
8.4
|
Eluční síla solventu vzorku je příliš silná oproti eluční síle mobilní fáze. Nekompatibilní solvent vzorku s mobilní fází
|
Frontující pík může mít tvar jak je ukázáno na obrázku. V případě frontujícího píku je třeba:
- rozpustit nebo naředit vzorek v mobilní fázi
- použít rozpouštědlo vzorku a slabší eluční síle
- nástřik vzorku o nižším objemu, přičemž platí, že při použití solventu o nižší eluční síle než má mobilní fáze, může se objem nástřiku vzorku pohybovat až kolem 10 % mrtvého objemu kolony, v případě použití solventu o vyšší eluční síle než má mobilní fáze, může se objem nástřiku vzorku pohybovat pouze kolem 1 % mrtvého objemu kolony. Vliv velikosti nástřiku na účinnost kolony
|
8.5
|
Velký mrtvý objem systému
|
Mezi kolonou a detektorem je zařazena kapilára o velkém mrtvém objemu (teoreticky). Dochází k rozšíření eluční zóny díky mimokolonovým příspěvkům. Pozor na zapojení kolony (obrázek).
|
|
Článek
|
Příčina
|
Korekce
|
9.1
|
Kontaminovaná nebo degradována kolona
nebo předkolona nebo kontaminován in-line filtr mobilní fáze.
|
Viz článek tabulky č. 1.2
|
9.2
|
Zařízení pro dávkování vzorku.
|
Zřejmě se jedná o netěsnost v nástřikovém zařízení – výměna těsnění jehly.
|
9.3
|
Nesprávné nastavení časové konstanty detektoru.
|
Viz článek tabulky č. 11.4
|
9.4
|
Příliš dlouhá cesta mezi kolonou a detektorem.
|
Zvyšuje se mrtvý objem systému, je nutné použít kratší kapiláru nebo kapiláru o menším vnitřním průměru.
|
9.5
|
Příliš vysoká nástřiková koncentrace
|
Snížit objem nástřiku nebo koncentraci vzorku. Chromatogram (pík) může mít takovýto tvar. (obrázek č. 10.1).
|
|
Článek
|
Příčina
|
Korekce
|
10.1
|
Použití IPC (chromatografie iontových
párů).
|
Pokud není vzorek rozpuštěn v mobilní fázi – rozpustit v mobilní fázi. V případě, že problém přetrvává, je nutné modifikovat mobilní fázi tak, aby nedocházelo k interferencím těchto píků s negativními píky.
|
10.2
|
Práce v blízké oblasti UV cutoff mobilní fáze – mobilní fáze má příliš vysokou absorbci.
|
Nastavit vlnovou délku nad UV cutoff mobilní fáze – práce ve vyšších oblastech vlnové délky.
|
10.3
|
Nesprávné nastavení změny vlnové délky u detektorů s programovatelnou vlnovou délkou.
|
Tento problém se vyskytuje při programování změny vlnové délky tam, kde dochází ke změně vlnové délky ještě na sestupné části píku. Je nutné posunout tuto změnu vlnové délky k vyšším retenčním časům. Viz obrázek č. 13.
|
10.4
|
Problém dávkování vzorku – nasátí vzduchové bublinky do jehly nebo dávkovací smyčky.
|
Propláchnout zařízení pro dávkování vzorku, jestliže problém přetrvává (autosampler), zkontroluje se, zda se netvoří vakuum při nasávání vzorku (příčinou je příliš pevné těsnění jehly kolem septa). Vyzkoušet nástřik vzorku z otevřené vialky bez septa nebo víčka.
|
|
Článek
|
Příčina
|
Korekce
|
11.1
|
Příliš vysoká nástřiková hmotnostní koncentrace (objem nástřiku /µl/ nebo koncentrace /µg/ml).
|
Při vysoké nástřikové hmotnosti se dostáváme do nelineární oblasti absorpční isotermy, chromatografická
kolona přestává separovat. Je nutné naředit vzorek nebo snížit objem dávkovaného vzorku na chromatografickou kolonu.
|
11.2
|
Na detektoru není elektronicky nastavena nula.
|
Na detektoru elektronicky nastavit nulu základní linie (Auto Zero).
|
11.3
|
Na detektoru nastavena příliš vysoká citlivost.
|
Snížit nastavení citlivosti detektoru.
|
11.4
|
Špatně nastavena časová konstanta
detektoru.
|
Nastavit správně časovou konstantu detektoru. Mimokolonové příspěvky k rozšíření chromatografické zóny
|
|
Článek
|
Příčina
|
Korekce
|
13.1
|
Vzorek degraduje
|
Viz článek tabulky 12.2
|
13.2
|
Ztráta účinnosti kolony – kolona degradována.
|
Viz článek
tabulky 1.2
|
13.3
|
Použitá jiná mobilní fáze.
|
Zkontrolovat použitou mobilní fázi. Je-li použitá mobilní fáze správná, použít čerstvě připravenou mobilní fázi.
|
13.4
|
Problém nástřiku vzorku na chromatografickou kolonu (autosampler):
- nízký nástřikový objem
- defektní jehla
- náběr vzorku mimo vialku
- malý objem vzorku ve vialce
|
Zkontrolovat zařízení pro nástřik vzorku. Při kontrole objemu nástřiku se postupuje tak, že se vialka naplní vodou (čistoty HPLC), zavíčkuje a zváží s přesností na 0,0001 g. Poté se z vialky odebere 6 krát 25 µl (mg) a opět se zváží se stejnou přesností. Rozdíl před a po náběru musí odpovídat 150 mg, v opačném případě je indikován problém nástřiku vzorku autosamplerem.
|
13.5
|
Problém nastavení detektoru:
- citlivost
- vynulování základní linie
- nastavení vlnové délky
- tok mobilní fáze mimo celu detektoru
- elektronický problém (defektní lampa)
|
Kontrola zapojení, nastavení detektoru, energie lampy – diagnostika detektoru.
|
13.6
|
Solvent vzorku je příliš viskózní.
|
Naředit vzorek mobilní fází pokud je toto možné, snížit rychlost nasávání vzorku.
|
13.7
|
Autosampler – porucha oplachu jehly.
|
Problém při oplachu jehly může být následující:
- defektní fluidika oplachu jehly
- porucha pumpy oplachu jehly
- kontaminovaný solvent oplachu jehly (vyměnit solvent, nepoužívat acetonitril)
- nasávání solventu oplachu jehly z rezervoáru samospádem
(snížit polohu rezervoáru, neměl by být výše než jehla)
|
13.8
|
Kontaminovaná kolona.
|
Viz článek tabulky 1.2
|
{kind=link}